When you pick up a generic pill at the pharmacy, you expect it to work just like the brand-name version. But how do manufacturers prove that? In the past, they relied on end-product testing-checking a few samples after production to see if they met specs. That approach was like locking the barn door after the horse escaped. Today, Quality by Design (QbD) has changed everything. It’s not about testing quality in at the end. It’s about building it in from day one.
What Is Quality by Design, Really?
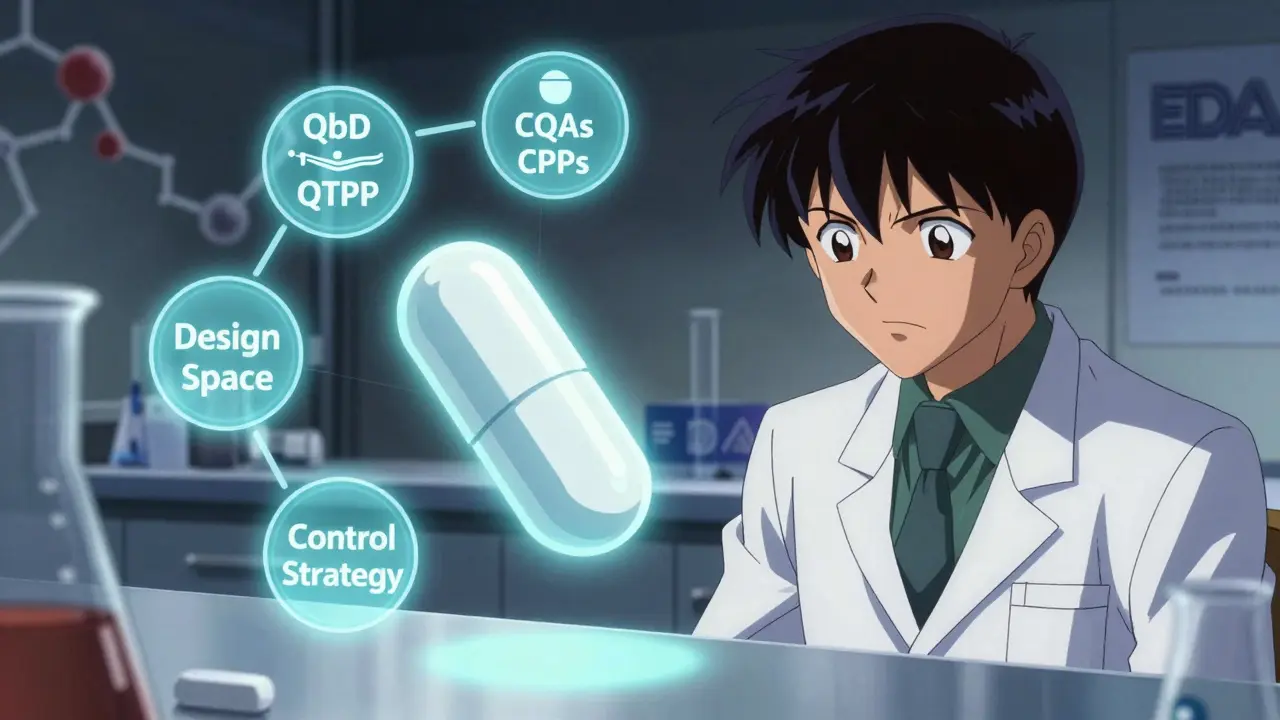
Quality by Design isn’t a buzzword. It’s a formal framework defined by the International Council for Harmonisation (ICH) Q8(R2). The idea is simple: start with the end goal in mind. What should the drug do? How should it behave in the body? What impurities are acceptable? These questions are answered upfront in the Quality Target Product Profile (QTPP). For generics, that means matching the reference drug-down to the dissolution rate, impurity levels, and how quickly it breaks down in the gut.
The FDA made QbD mandatory for all Abbreviated New Drug Applications (ANDAs) submitted after October 1, 2017. That’s not a suggestion. It’s a requirement. And it’s working. Since then, approval rates for generic applications have jumped 23%, and review times dropped by nearly five months on average. Why? Because regulators aren’t guessing anymore. They’re seeing the science.
The Five Pillars of QbD in Generic Development
QbD isn’t one step. It’s five interconnected pieces that work together.
- Quality Target Product Profile (QTPP) - This is your blueprint. For a generic tablet, it includes identity, strength, dissolution profile, and impurity limits. The FDA requires at least 95% similarity to the brand-name drug’s performance. If your dissolution curve doesn’t match the reference drug within tight limits, the application gets rejected.
- Critical Quality Attributes (CQAs) - These are the measurable characteristics that affect safety and effectiveness. For most generics, developers identify 5 to 12 CQAs. The big ones? Dissolution (f2 similarity factor >50), content uniformity (RSD under 6%), and impurity profiles (following ICH Q3B thresholds). Miss one, and you risk a Complete Response Letter.
- Critical Process Parameters (CPPs) - These are the manufacturing settings that directly impact your CQAs. Think granulation moisture (1.5-3.0%), compression force (10-15 kN), drying temperature (40-50°C). You don’t just pick a number. You test ranges using Design of Experiments (DoE) to find what works.
- Design Space - This is the game-changer. Instead of one fixed setting, you define a multidimensional zone where all parameters still produce a quality product. The FDA accepts design spaces built on data from 100+ simulated batches, with 95% confidence that every batch will meet specs. That means you can tweak your process without asking for approval every time.
- Control Strategy - How do you know you’re staying in the design space? That’s where Process Analytical Technology (PAT) comes in. Near-infrared spectroscopy, Raman probes, and real-time moisture sensors let you monitor production as it happens. Companies using PAT reduce end-product testing by 35-60%, cutting costs and speeding up release.
Why QbD Beats the Old Way
Traditional generic development was like baking cookies with a fixed recipe: mix for 15 minutes at 25°C, bake for 12 minutes. If one batch failed, you blamed the batch-not the process. QbD flips that. You test variables to understand how they interact. That’s why QbD-based products are 28-42% more robust during scale-up, according to a Tufts CSDD study of 127 generics.
The numbers speak for themselves:
- QbD applications get 31% fewer Complete Response Letters from the FDA.
- Approval timelines average 9.2 months vs. 13.9 months for non-QbD.
- Manufacturers save $1.2-2.8 million per product annually by avoiding costly change notifications.
- Process changes happen 73% faster when you have an approved design space.
Take Teva’s levothyroxine case. By using QbD and continuous manufacturing, they improved batch consistency by 28%. That’s not luck. That’s science.

The Hidden Costs and Real Challenges
QbD isn’t free. It’s expensive upfront. Development timelines stretch by 4-8 months. Training scientists in risk management and DoE costs 80-120 hours per person. PAT equipment? Minimum $500,000. Software like MODDE Pro? $15,000 per user per year.
And not every product needs it. For a simple immediate-release tablet with a well-known formulation, over-engineering QbD can be wasteful. Dr. James Polli from the University of Maryland points out cases where companies spent $450,000 on DoE studies for products that didn’t need it. The key is proportionality. Don’t use a sledgehammer to crack a nut.
Biggest hurdles?
- Establishing in vitro-in vivo correlation (IVIVC) for complex products like extended-release tablets. 22% of applicants fail here, per EMA data.
- Justifying design space boundaries for multi-component formulations. 41% of manufacturers struggle with this.
- Cost pressure. Indian generics firms adopt QbD at 68%-lower than U.S. or EU firms-because budgets are tight. But the top 10 Indian companies still invested $227 million in QbD capabilities in 2022. They know the long-term payoff.
How the Industry Is Evolving
QbD adoption is accelerating. In 2018, only 38% of ANDAs included QbD elements. By 2022, that jumped to 74%. For complex generics-like inhalers, patches, or injectables-it’s nearly 92%. The FDA’s QbD Pilot Program processed 87 submissions with a 92% first-cycle approval rate. Compare that to 78% for traditional submissions.
New guidelines are pushing it further. ICH Q14 (Analytical Procedure Development), effective December 2023, requires more robust method validation-but rewards it with 40% faster approval. The FDA’s Emerging Technology Program has approved all 27 QbD-based continuous manufacturing applications it’s seen.
What’s next? 3D-printed generics. Complex biologics follow-ons. The FDA’s 2024-2026 plan targets these. By 2027, McKinsey predicts 95% of new generic approvals will use QbD.

Best Practices That Actually Work
Companies that succeed with QbD follow three proven strategies:
- Use the reference drug smartly. Don’t reinvent the wheel. Leverage advanced analytical techniques to characterize the brand-name product. This cuts development time by 30%.
- Bracket multi-strength products. Instead of testing every strength, test the extremes (highest and lowest dose). If those work, the middle ones likely do too. This reduces studies by 45%.
- Go continuous. Continuous manufacturing, paired with QbD, improves consistency and reduces batch-to-batch variation. Teva’s case proves it.
Dr. Elena Rodriguez at Hikma Pharmaceuticals saw post-approval deviations drop from 14 to 2 per year after switching to QbD. That saved $850,000 annually in investigations. But it wasn’t easy. Her team spent 120 person-hours on training. The first two submissions were rough. But once they got the system right, things stabilized.
Is QbD Right for Your Product?
Ask yourself:
- Is this a simple immediate-release tablet? Maybe skip heavy DoE. Focus on dissolution and uniformity.
- Is it a complex delivery system-patch, inhaler, injectable? QbD isn’t optional. It’s essential.
- Do you plan to make 10 strengths? Use bracketing. Save time and money.
- Is your product under $50 million in annual revenue? Be careful. Don’t let QbD costs eat up more than 15% of projected lifetime revenue.
The goal isn’t to do the most science. It’s to do the right science. QbD gives you flexibility, predictability, and regulatory trust. But only if you apply it with purpose.
What is the main difference between traditional generic development and QbD?
Traditional development relies on fixed manufacturing recipes and end-product testing to ensure quality. QbD starts with defining desired product performance upfront, then uses scientific data to identify how process variables affect quality. Instead of testing each batch, you control the process within a proven design space, making quality predictable and consistent.
Why does the FDA require QbD for generic drugs?
The FDA requires QbD to ensure that generic drugs are consistently safe and effective, not just by matching labels but by matching performance. QbD reduces reliance on random sampling and increases confidence that every batch will work the same way. It also gives regulators transparency into how the product is made, reducing review times and approval delays.
Can QbD help reduce manufacturing costs over time?
Yes. While QbD increases upfront costs, it lowers long-term expenses. Companies with approved design spaces can make process changes without submitting new applications, saving $1.2-2.8 million per product annually. Reduced batch failures, fewer regulatory queries, and faster releases also cut operational costs. Teva and Hikma reported annual savings of hundreds of thousands to over $800,000 after implementing QbD.
Do I need expensive equipment to implement QbD?
You don’t need everything at once, but key tools like PAT (Process Analytical Technology) are critical for modern QbD. Near-infrared spectroscopy, Raman probes, and real-time moisture sensors help monitor processes during production. The minimum investment for PAT equipment is around $500,000. Software for multivariate analysis (like MODDE Pro) adds another $15,000 per user per year. But these tools reduce end-product testing by 35-60%, which offsets the cost.
Is QbD only for large pharmaceutical companies?
No. While large firms lead adoption, mid-sized and even smaller companies are using QbD strategically. The key is proportionality. A small company making a simple tablet can use a streamlined QbD approach-focusing on critical parameters without full DoE. Regulatory agencies like the FDA offer free training modules and pilot programs to help smaller players get started. The goal isn’t to replicate big pharma’s approach, but to apply the right level of science for your product.
How does QbD affect bioequivalence testing?
QbD doesn’t eliminate bioequivalence testing, but it reduces the need for clinical trials. By establishing strong in vitro dissolution profiles and proven design spaces, manufacturers can demonstrate equivalence through lab testing alone. The FDA accepts dissolution data with f2 similarity >50 as sufficient for many immediate-release generics. For complex products, IVIVC models are used to link in vitro results to in vivo performance, making clinical studies unnecessary in many cases.






Pawan Chaudhary
December 16, 2025 AT 08:09This is such a refreshing take on generics! I work in pharma in India, and seeing QbD finally get the attention it deserves makes me proud. The cost fears are real, but the long-term wins? Totally worth it. We’re seeing fewer recalls and happier regulators now. 🙌
Jonathan Morris
December 18, 2025 AT 06:03Let’s be honest - this whole QbD push is just a bureaucratic power grab disguised as science. The FDA doesn’t care about ‘predictability’ - they care about control. Every time a manufacturer tweaks a process, they’re forced to submit a supplemental application anyway. This is regulatory theater with extra steps.
Donna Packard
December 18, 2025 AT 07:30I appreciate how this breaks down the science without oversimplifying. It’s easy to think of generics as ‘cheap copies,’ but this shows how much precision actually goes into them. The design space concept alone is brilliant - it’s like giving manufacturers room to breathe while keeping patients safe.
Patrick A. Ck. Trip
December 18, 2025 AT 22:21While I’m not a scientist, I’ve read a lot on this topic. The QbD framework seems like a logical evolution - though I wonder if the cost barriers might unintentionally limit access to generics in lower-income regions. Maybe the FDA could offer subsidized PAT training or open-source design space templates?
Sam Clark
December 19, 2025 AT 21:14This is a well-structured and evidence-based overview. The data on approval timelines and cost savings is compelling. However, I would emphasize that QbD implementation must be scaled appropriately to product complexity. Over-application risks inefficiency, while under-application risks non-compliance. Balance is key.
amanda s
December 21, 2025 AT 00:58So now we’re forcing American companies to spend half a million dollars on sensors just so Indian manufacturers can undercut them with cheaper generics? This isn’t science - it’s economic warfare disguised as regulation. The FDA is letting foreign labs dictate our standards.
Sachin Bhorde
December 21, 2025 AT 18:35Bro, QbD is legit. We did a DoE study on a 500mg metformin tab last year - turned out the granulation moisture was the real MVP. We saved 2 months on approval and cut waste by 30%. PAT gear is pricey, yeah, but once you’re in, you never go back. Also, MODDE Pro? Worth every rupee. #QbDlife
Joe Bartlett
December 23, 2025 AT 02:58QbD is just common sense. Test early, fix early. Why did it take 20 years to get here? Simple products don’t need fancy sensors. Complex ones do. Done.
Jody Patrick
December 23, 2025 AT 22:11QbD is just another way for regulators to make life harder for small manufacturers. We’re not Big Pharma. We don’t have $500k to throw at a Raman probe.
Radhika M
December 25, 2025 AT 13:16My cousin works at a generic lab in Mumbai. They started using QbD last year. Now their approval rate is 90%. Before? Barely 50%. It’s not magic - it’s just better planning. Also, they saved money on retests. Win-win.
Philippa Skiadopoulou
December 25, 2025 AT 22:55The shift from end-product testing to process control represents a paradigm change in pharmaceutical quality assurance. The regulatory alignment between ICH Q8(R2) and FDA guidance is commendable. Implementation challenges remain, particularly regarding IVIVC establishment.
Chris Van Horn
December 26, 2025 AT 08:54Oh, so now we’re supposed to believe that a $15,000 software license makes a generic drug ‘better’? This is why American healthcare is broken. You spend millions on sensors and software just to make a pill that’s chemically identical. Meanwhile, people can’t afford insulin. This isn’t innovation - it’s corporate theater.
Virginia Seitz
December 27, 2025 AT 18:51So cool! 🤩 I had no idea generics were this scientific now. My grandma takes them every day - now I can tell her they’re basically engineered for perfection. 🙏💊
Peter Ronai
December 27, 2025 AT 19:00Everyone’s acting like QbD is some revolutionary breakthrough. Newsflash: the science has existed since the 90s. What changed? The FDA finally realized they could use it to delay approvals under the guise of ‘rigor.’ This isn’t progress - it’s regulatory obstruction with a fancy acronym.
Steven Lavoie
December 29, 2025 AT 09:44I’ve seen this play out in both U.S. and EU labs. The cultural shift is real - engineers are now trained alongside pharmacologists. That collaboration is what makes QbD work. It’s not just tools or software. It’s people thinking differently. And that’s the real win.